
Chap.13 计算小白硬学VASP —— 自动搜索表面吸附位点并生成吸附结构
¡Hola a todos!
本章介绍如何在slab模型表面自动搜索吸附位点并生成吸附模型。
AdsorbateSiteFinder搜索吸附位点
AdsorbateSiteFinder模块主要用于搜索slab模型表面潜在的吸附位点,主要分为三大类:ontop、bridge、hollow。首先导入AdsorbateSiteFinder模块:
1 | from pymatgen.analysis.adsorption import AdsorbateSiteFinder |
slab模型以Si(111)面为例,关于如何自动切slab模型可以参考往期内容。
模型:
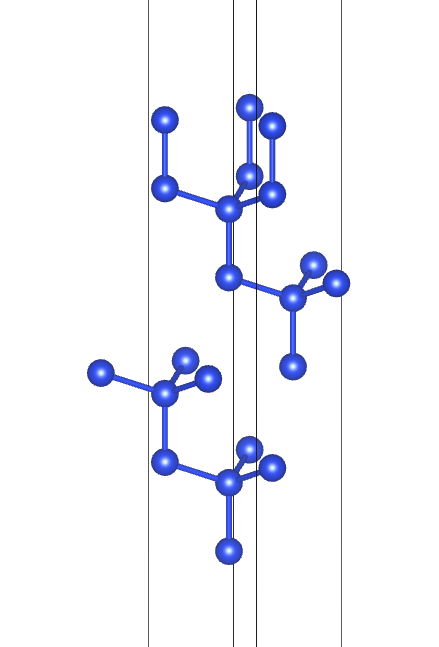
1 | from pymatgen.analysis.adsorption import AdsorbateSiteFinder |
调用find_adsorption_sites()方法搜索吸附位点,find_adsorption_sites()默认的吸附距离为2埃。可以传入distance参数进行覆盖
1 | from pymatgen.analysis.adsorption import AdsorbateSiteFinder |
运行代码,可以得到所有潜在吸附位点的坐标:
1 | {'all': [array([ 3.36811902, 0.27779776, 28.32198049]), |
初次之外,还可以导入plot_slab()方法实现可视化,代码如下:
1 | from pymatgen.analysis.adsorption import AdsorbateSiteFinder, plot_slab |
plot_slab()主要需要传入三个参数:
slab:表面模型ax:matplotlib对象用于绘图adsorption_sites:是否显示吸附位点标识
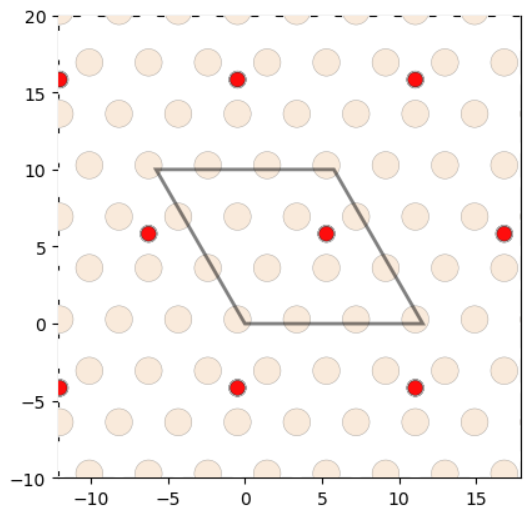
运行代码后可得到:
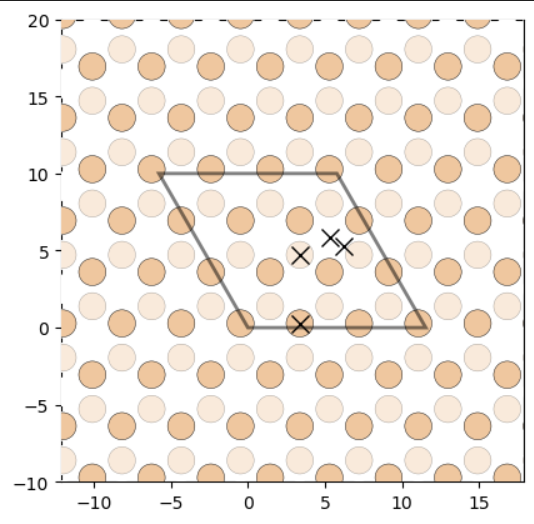
深色表示最表层原子,浅色表示次表层原子;平行四边形框表示3x3x1超胞的Si_111结构,X表示对应的潜在吸附位点。
添加吸附原子/分子
搜索完吸附位点后,下一步需要给slab增加吸附的原子/分子,最直接的方法为直接写对应的原子分子坐标,然后传入,以吸附CO为例,代码如下:
1 | from pymatgen.core.structure import Structure |
首先,该方法先要导入Molecule模块,Molecule仅需要传入,原子元素种类及对应的坐标,且一一对应。这里传入的坐标对应的空间与slab模型无关,可以认为是在一个空盒子里面构建分子。
然后,调用AdsorbateSiteFinder模块的add_adsorbate()方法把吸附分子加在slab上。
add_adsorbate()需要读取分子结构molecule=CO,以及吸附坐标ads_coord=adsorption_sites['hollow'][0],这里坐标选取第一个hollow位。
运行代码:
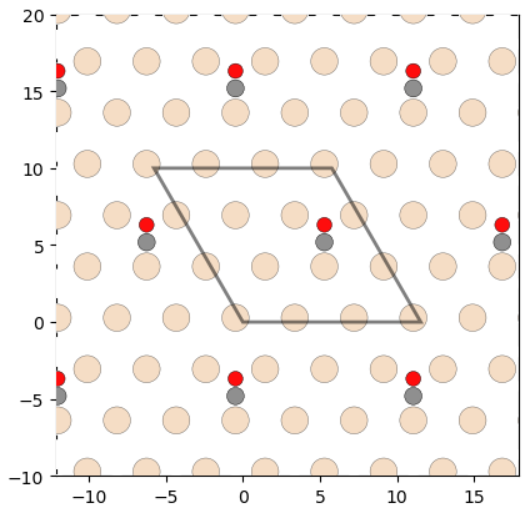
结果显示CO的吸附情况,可以导出cif结构查看:
1 | ...... |
VESTA查看:
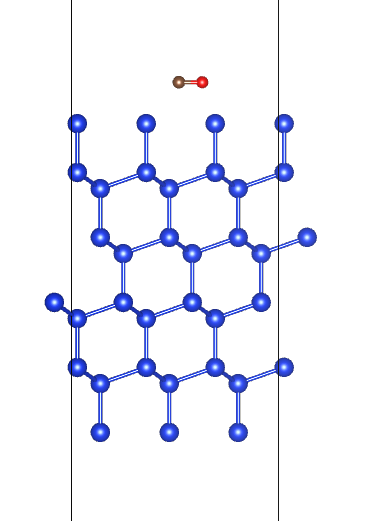
进阶用法
上述操作中,如果每次吸附分子都需要自己写坐标的话,确实会比较麻烦。对于复杂的分子,自己写坐标的难度也比较大。
因此,这里推荐调用ase来自动导入分子结构,主要用到的模块为:
1 | from ase.build.molecule import molecule |
ase的molecule模块支持输入常见的分子化学式就可以构建对应的分子结构,可以通过如下代码查看支持的分子种类:
1 | from ase.build.molecule import extra |
运行代码:
1 | ['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF', 'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2', 'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3', 'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane', 'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2', 'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl', 'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN', 'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO', 'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2', 'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C', 'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S', 'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d', 'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O', 'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF', 'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4', 'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O', 'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO', 'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl', 'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2'] |
Note:ase得到的分子结构不能直接使用,还需要转化成pymatgen可以读取的格式,需要用到AseAtomsAdaptor,完整代码如下:
1 | from ase.build.molecule import molecule |
运行代码:
导出cif文件在VESTA查看:
1 | ...... |
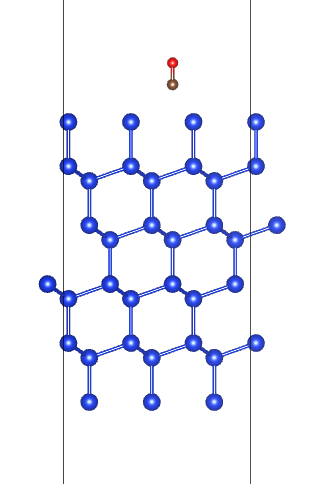
Note:对比两种方法可以发现,ase中的CO分子默认是垂直吸附。该方法的缺点即无法自定义吸附姿态,如果需要自定义吸附姿态,还是需要自己写坐标,或者其他软件建模导入。
¡Muchas gracias!
- 标题: Chap.13 计算小白硬学VASP —— 自动搜索表面吸附位点并生成吸附结构
- 作者: 炫酷老司机
- 创建于 : 2024-08-07 00:00:00
- 更新于 : 2024-08-07 10:04:46
- 链接: https://andyhox.github.io/2024/08/07/Learn-VASP-from-pymatgen-13/
- 版权声明: 欢迎个人转载、使用、转贴等,但请获得作者同意且注明出处!